
Help!
MicRhoDE provides the scientific community with a comprehensive, high-quality and freely accessible resource of nucleic acid sequences encoding prokaryotic rhodopsins, with an emphasis on proteorhodopsins.
If you need complementary information about any point discussed in the following sections, please contact us.
Documentation
• Sequence retrieving & cleaning strategies :
MicRhoDE is based on an extensive worldwide Illumina™ sequencing of marine proteorhodopsin diversity (PRGD) (see the MicRhoDE flowchart below). In addition, a set of reference sequences found in literature was added to constitute a complete and diversified seed for BLAST sequence similarity search. Sequences from GenBank or GOS databases were retrieved using different search strategies (BLASTn, BLASTp, tBLASTn) and loosed similarity threshold.
Blast results were dereplicated, manually checked for quality and consistency (is the predicted protein sequence consistent regarding known rhodopsins?), and trimmed to identify open reading frames using bacterial genetic code (11).• Alignment strategy :
Since the prokaryotic rhodopsins are membrane embedded proteins constituted by 7 alpha-helixes, evolutionary constrains vary according to the protein region. As a consequence, putative structure of the protein has to be considered when nucleic acid sequences are aligned. Since aligning 7,857 nucleic acid sequences according to the secondary structure of the corresponding proteins is time-consuming, the sequence set has been separated in two. The 3,871 longest sequences (>100 amino acid residues) were aligned according to the protein secondary structure using MAFFT eINSi strategy. The 3,986 shortest ones were added to the robust alignment using MAFFT FFT strategy with the --addfragments option that conserves the original alignment.
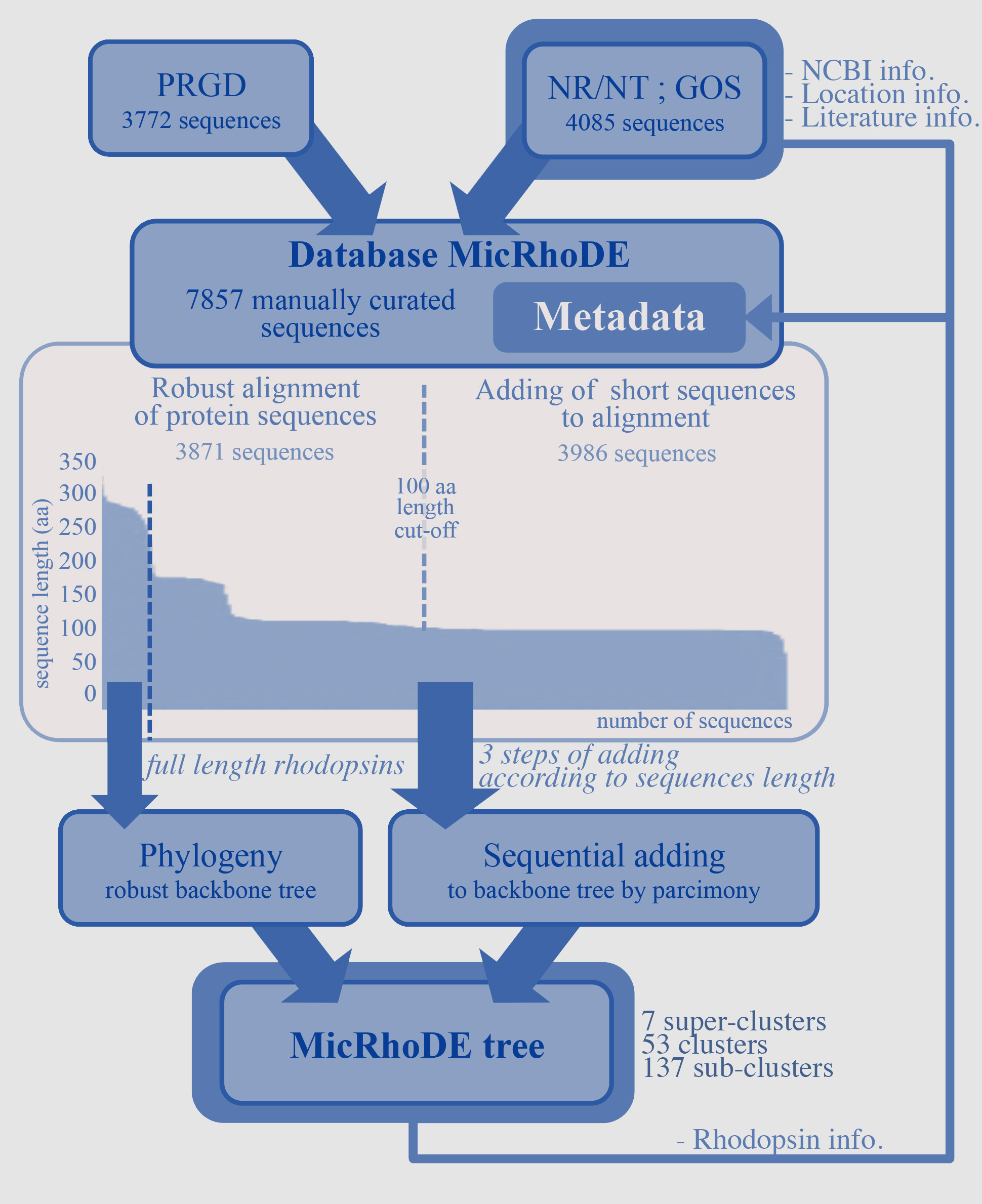
MicRhoDE flowchart
• Phylogenetic tree construction strategy :
• Metadata assignation strategy :
How to
• Search a specific information/sequence ?
In the "Search" tab, users can extract supercluster, cluster, or subcluster sequences from the database, and/or selected predicted spectral tuning, putative activity and function, and/or biome of isolation (ie seawater, freshwater or terrestrial), and/or marine province of isolation. Searches by keyword are also possible using "string search".
Select metadata you wish associate to the results and then select your output preferences as tables or fasta sequences or visualize the results in a map. By using the latter option, isolation sites of matching sequences are mapped. By clicking on a geographical site, you can see and download all the sequences retrieved from it.
Click on "Submit" button to search or "Reset" to clean all fields.
• Blast your sequence ?
In the "Blast" tab, you may submit your nucleic acid or protein sequence and search for similar sequences in MicRhoDE. Enter your query, choose of the blast type you wish to use : you can perform a BLASTn if you submit a nucleic acid sequence, a BLASTp if you submit a protein sequence, a tBLASTn if you submit a protein sequence and wish to search for similar nucleic acid sequences translated into proteins, and a tBLASTx if you submit a nucleic acid sequence and wish to search the corresponding translated protein sequence for similar nucleic sequences translated into proteins. You can also blast your query against the entire MicRhoDE database or only against cultivated strains.
Select your metadata and output preferences as indicated above.
Click on "Submit" button to search or "Reset" to clean all fields.
• Place your sequence into the MicRhoDE phylogenetic tree ?
In the "Phylogeny" tab, you may place your sequence into the reference phylogenetic tree. "Go to the placer" to use the MicRhoDE workflow. First upload your sequence (STEP1) and then execute the MicRhoDE workflow (STEP2) as described on the left panel. Then follow the description of MicRhoDE workflow as indicated in the central pannel. The results (trees) can be viewed in the right pannel by cliking on the little eye (view data).
• Explore the MicRhoDE phylogenetic tree ?
In the "Phylogeny" tab are displayed 2 views of the phylogenetic tree: a compressed one showing only superclusters and a complete and browsable one (below). For a more advanced use, a newick version of the tree and the ARB version of the whole database are both available on the download page.
• Explore the biogeography ?
In the "Map" tab, an interactive map is displayed. You can visualize the data using satellite, plan or relief views and navigate. The red flags indicate locations for which data are available. Only 843 (10.7%) sequences do not have information about their geographical origin. The number in red flags indicates the number of sequences available for this location. If you click on a flag, a pop-up window will inform you about the hexadecimal GPS position, the number of sequences, the number and the dominance of superclusters and clusters, and the proportion of predicted spectral variants.
